
The pharmaceutical industry operates within a complex regulatory framework where two critical applications determine a drug product’s journey from laboratory concept to patient treatment. The investigational new drug (IND) and new drug application (NDA) processes represent distinct regulatory phases that every drug developer must navigate successfully. These applications serve fundamentally different purposes within the drug development continuum, yet confusion between their requirements, timelines, and strategic implications continues to challenge even experienced pharmaceutical companies.
Understanding these distinctions becomes particularly crucial as the United States maintains stringent oversight of new drug development through the Food and Drug Administration. The consequences of misunderstanding these regulatory pathways extend beyond delayed timelines—they can determine the ultimate success or failure of promising therapeutic interventions.
Each application type demands specific expertise, documentation standards, and strategic planning that directly impact development costs and market entry potential.

Understanding IND Applications
An investigational new drug application represents the formal request that enables clinical testing of experimental therapies in human subjects within the United States. This application serves as the regulatory gateway between preclinical research and human clinical trials, establishing the legal framework for administering unapproved drug products to study participants.
The IND application process focuses primarily on safety assessment rather than efficacy demonstration. Drug developers must provide comprehensive preclinical data, manufacturing information, and detailed clinical protocols that demonstrate reasonable safety for proposed human studies. The application includes animal pharmacology and toxicology data, manufacturing specifications, and clinical trial protocols with investigator qualifications.
A critical component of the IND process involves the clinical hold mechanism, which grants regulatory authorities the power to delay or suspend clinical trials when safety concerns arise. This regulatory tool ensures that clinical trial participants receive adequate protection while allowing promising therapies to advance through development phases. Drug developers must understand that clinical hold decisions can significantly impact development timelines and require immediate responsive action.
The IND application remains active throughout the clinical development process, requiring regular safety updates, protocol amendments, and annual reports. This ongoing regulatory relationship establishes the foundation for eventual marketing approval applications while maintaining continuous safety oversight of investigational therapies.
NDA Fundamentals
The new drug application represents the comprehensive submission seeking marketing approval for commercial distribution of a drug product in the United States. Unlike IND applications that focus on clinical investigation authorization, NDAs demonstrate that sufficient evidence exists to support both safety and effectiveness claims for specific therapeutic indications.
NDA submissions require extensive clinical data from well-controlled studies that provide enough evidence of therapeutic benefit to justify commercial approval. This includes Phase I, II, and III clinical trial results, comprehensive safety databases, risk assessment evaluations, and proposed labeling that accurately reflects the drug’s benefit-risk profile.
The drug evaluation process for NDAs involves rigorous scientific review by multidisciplinary teams examining clinical efficacy, safety profiles, manufacturing quality, and proposed labeling. Standard review timelines extend 10-12 months, though priority review designations can reduce this timeframe to 6-8 months for therapies addressing unmet medical needs.
Manufacturing documentation within NDA submissions must demonstrate consistent production capability, quality control systems, and supply chain reliability necessary for commercial distribution. This includes detailed chemistry, manufacturing, and controls information that ensures product quality and patient safety throughout the commercial lifecycle.
Risk evaluation and mitigation strategies may be required as part of NDA approval, particularly for drug products with significant safety concerns that require specialized monitoring, distribution restrictions, or healthcare provider education programs.
Critical Distinctions Analysis
The fundamental distinction between IND and NDA applications lies in their regulatory objectives: investigation versus commercialization. IND applications seek permission to study experimental therapies, while NDAs request authorization for commercial marketing based on demonstrated clinical benefit.
Evidence requirements differ substantially between these application types. IND submissions focus on preclinical safety data sufficient to justify human exposure, whereas NDAs require comprehensive clinical evidence demonstrating both safety and effectiveness across diverse patient populations. The evidentiary threshold for NDA approval significantly exceeds IND requirements, reflecting the transition from controlled research settings to broad clinical practice.
Timeline considerations create strategic implications for drug development planning. IND applications typically receive responses within 30 days, enabling rapid clinical trial initiation when safety data support human testing. Conversely, NDA review processes require extensive evaluation periods that demand careful resource allocation and market entry planning.
Cost implications vary dramatically between application types. IND submissions involve relatively modest regulatory fees and documentation requirements, while NDA applications require substantial user fees, extensive manufacturing data, and comprehensive clinical documentation that represents a significant investment in regulatory preparation.
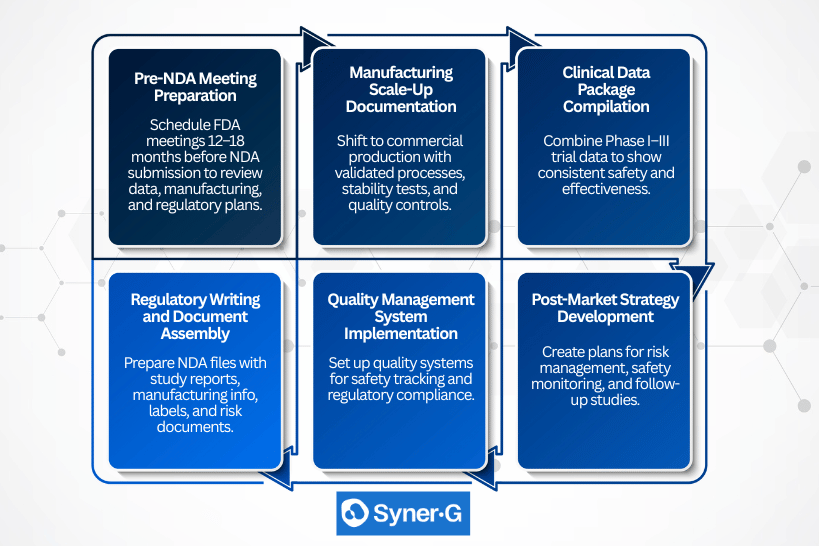
Transitional Steps Between IND and NDA
The transition from investigational new drug status to new drug application represents a critical inflection point in drug development that requires systematic planning and execution. This regulatory evolution demands careful coordination between clinical, manufacturing, and regulatory teams to ensure seamless progression from clinical investigation to commercial readiness.
The transitional phase typically begins during late-stage clinical development when enough evidence accumulates to support marketing approval considerations. Drug developers must anticipate NDA requirements throughout the clinical trial process, ensuring data collection standards meet commercial approval thresholds while maintaining IND compliance obligations.
Essential Steps for IND-to-NDA Transition:
- Pre-NDA Meeting Preparation – Schedule FDA meetings 12-18 months before planned NDA submission to discuss clinical data requirements, manufacturing expectations, and regulatory pathway optimization.
- Manufacturing Scale-Up Documentation – Transition from clinical-grade to commercial-scale manufacturing with comprehensive process validation, stability studies, and quality control system implementation.
- Clinical Data Package Compilation – Aggregate Phase I-III clinical trial results into integrated safety and efficacy databases that demonstrate consistent benefit-risk profiles across study populations.
- Regulatory Writing and Document Assembly – Develop comprehensive NDA modules including clinical study reports, manufacturing information, proposed labeling, and risk assessment documentation.
- Quality Management System Implementation – Establish commercial-grade quality systems that support post-market surveillance, adverse event reporting, and ongoing safety monitoring requirements.
- Post-Market Strategy Development – Create risk evaluation and mitigation strategies, pharmacovigilance plans, and post-market study commitments that address regulatory concerns identified during development.
The formal request process for NDA submission requires substantial resource allocation and strategic timing considerations that align with market entry objectives and competitive landscape dynamics within the United States pharmaceutical market.
Related Article: The Evolution of US Biologic Drug Regulations
Strategic Implementation
Successful navigation of both regulatory pathways requires early planning that anticipates IND-to-NDA transition requirements. Drug developers should establish regulatory strategies that align IND clinical development with eventual NDA submission requirements, ensuring clinical trial designs generate data suitable for marketing approval applications.
Common pitfalls include inadequate manufacturing documentation during IND phases that creates NDA submission challenges, insufficient safety database development that delays marketing approval, and clinical trial designs that fail to address regulatory requirements for effectiveness demonstration.
Success factors include early regulatory consultation through FDA meetings, a comprehensive understanding of target indication requirements, and integrated development planning that considers both investigational and commercial regulatory needs throughout the drug development process.
Overview of Related FDA Drug Applications (BLA, ANDA, OTC)
The FDA drug approval ecosystem extends beyond IND and NDA applications to encompass specialized regulatory pathways designed for specific product categories and development scenarios. Understanding these related applications provides drug developers with comprehensive regulatory strategy options that may better align with particular therapeutic approaches or commercial objectives.
Biologics License Applications
Biologics License Applications (BLA) govern the approval process for biological products, including vaccines, blood products, and complex biotechnology-derived therapeutics. BLA requirements parallel NDA standards but incorporate additional considerations for manufacturing complexity, immunogenicity assessment, and product characterization unique to biological drug products. The clinical trial requirements for BLA submissions often demand specialized expertise in biologics development and regulatory science.
Abbreviated New Drug Applications
Abbreviated New Drug Applications (ANDA) provide the regulatory pathway for generic drug products seeking to demonstrate bioequivalence to previously approved reference drugs. ANDA submissions focus primarily on pharmaceutical equivalence and bioequivalence rather than clinical efficacy demonstration, creating accelerated approval timelines for established therapeutic entities. This pathway significantly reduces drug development costs and timelines compared to traditional new drug application processes.
Over-the-Counter Drug Applications
Over-the-counter (OTC) drug applications address consumer products available without prescription requirements. The OTC regulatory framework includes monograph compliance for established ingredients and New Drug Applications for novel OTC formulations. Recent regulatory modernization efforts have streamlined OTC pathways while maintaining consumer safety standards through updated labeling requirements and post-market surveillance systems.
Combination product applications require coordination between drug, device, and biologics regulatory centers when therapeutic products incorporate multiple regulated components. These applications demand integrated development strategies that address each component’s regulatory requirements while demonstrating overall product safety and effectiveness.
Orphan drug designations can apply across multiple application types, providing regulatory incentives for rare disease therapeutics regardless of whether they follow traditional NDA, BLA, or specialized approval pathways. These designations offer market exclusivity, fee waivers, and enhanced regulatory support that can significantly impact commercial viability for niche therapeutic markets.
The strategic selection among these regulatory pathways requires careful consideration of product characteristics, target patient populations, competitive landscapes, and commercial objectives that align with specific drug development goals and market entry strategies.

Regulatory Trends and Future of IND/NDA Processes
The regulatory landscape for drug development continues evolving through technological advancement, scientific innovation, and policy modernization initiatives that reshape traditional IND and NDA paradigms. Contemporary trends emphasize accelerated pathways, digital transformation, and patient-centric approaches that fundamentally alter how drug developers approach regulatory strategy.
Real-world evidence integration represents a significant trend affecting both investigational new drug applications and new drug applications. Regulatory authorities increasingly accept real-world data to supplement traditional clinical trial evidence, particularly for rare diseases and post-market safety monitoring. This evolution enables more flexible clinical trial designs and potentially reduces development timelines for certain therapeutic areas.
Digital health technologies and artificial intelligence applications are transforming drug evaluation processes within regulatory frameworks. Electronic submissions, predictive analytics for safety signal detection, and automated data review systems enhance regulatory efficiency while maintaining scientific rigor. These technological advances create opportunities for streamlined IND applications and more sophisticated NDA review processes.
Expedited pathway utilization continues expanding beyond traditional breakthrough therapy and fast track designations. Regulatory authorities develop new mechanisms for addressing unmet medical needs, including expanded access programs, conditional approvals, and adaptive clinical trial designs that blur traditional boundaries between investigation and commercialization phases.
International harmonization efforts influence domestic regulatory processes as global drug development strategies require coordination across multiple regulatory jurisdictions. The clinical trial phase alignment and manufacturing standards convergence create efficiencies that benefit drug developers pursuing marketing approval in multiple markets simultaneously.
Patient engagement integration throughout drug development processes represents an emerging trend that affects both clinical investigation design and marketing approval considerations. Regulatory authorities increasingly incorporate patient perspectives into benefit-risk assessments, potentially influencing approval decisions for therapies addressing significant unmet medical needs.
Turning Regulatory Knowledge Into Competitive Advantage
The distinction between IND and NDA applications represents more than regulatory technicality—it defines the strategic framework for successful drug development. Understanding these critical differences enables informed decision-making that accelerates therapeutic innovation while ensuring patient safety and regulatory compliance throughout the development lifecycle.

{kind=link}