
Your Phase 1/2 data is striking, the safety profile is clean, and the board is asking how quickly you can get to market. Breakthrough therapy designation is often used as shorthand for valuation and speed, but the leap from promising data to an official FDA designation takes more than high percentages.
The FDA defines BTD as a process to expedite the development and review of drugs for serious conditions where preliminary clinical evidence suggests the drug may demonstrate substantial improvement over existing therapies on at least one clinically significant endpoint.
This guide covers specific eligibility criteria, the benefits of intensive FDA guidance, and the mechanics of the formal request. We also compare BTD to Fast Track and Priority Review while addressing hidden risks in CMC and confirmatory evidence that often catch lean teams off guard.
Talk to our regulatory team about your BTD strategy.
Breakthrough Therapy Designation: A Development Accelerator, Not a Lowered Bar
Is breakthrough therapy designation a guaranteed shortcut to market? Contrary to common belief, BTD is an intensive engagement model rather than a lowered approval standard. The FDA created it to expedite development of drugs for serious conditions where preliminary clinical evidence indicates a potential for substantial improvement over available therapies on at least one clinically significant endpoint.
BTD does not reduce evidentiary requirements for safety or efficacy. It also does not guarantee Priority Review or Accelerated Approval, though these pathways often stack. Instead, BTD functions as a high-touch partnership within the FDA’s suite of expedited programs for serious conditions.
So what exactly must be true to qualify for this level of agency collaboration?
Eligibility Criteria for Breakthrough Therapy Designation
Consider a biotech presenting strong Phase 1 data for a rare oncology target, only to realize their “substantial improvement” claim relies on a surrogate endpoint the FDA has not yet validated. To determine if a program falls in the BTD “plausible zone,” sponsors must evaluate evidence against a two-part checklist:
- Serious or Life-Threatening Condition: The FDA focuses on conditions with significant morbidity or high mortality risk, such as progressive diseases that impact survival or daily functioning.
- Preliminary Clinical Evidence: This requires human clinical data. Preclinical narratives or animal models are insufficient. The data must show a clear advantage over available therapies on at least one clinically significant endpoint.
A common sponsor mistake is confusing “unmet medical need” (Fast Track criteria) with “substantial improvement” (BTD criteria). Proving substantial improvement the way the FDA expects is usually the hardest part.

Framing the Evidence: Endpoint Selection and the Comparator Trap
Roughly 60% of BTD requests are denied or withdrawn each year. These rejections often happen because “striking” clinical data fails to meet the FDA’s specific definition of a clinically significant benefit. Success requires aligning the magnitude and duration of effect with the clinical importance of the outcome.
The FDA evaluates endpoints based on several rigorous criteria:
- Irreversible Morbidity or Mortality (IMM): The gold standard for clinical significance.
- Serious Symptoms: Direct relief of the disease burden.
- Surrogate/Intermediate Endpoints: These must be “reasonably likely” to predict clinical benefit.
- Safety: A significantly improved profile may be sufficient if efficacy is comparable to existing treatments.
The Available Therapy Problem
Sponsors must also manage the “available therapy” trap — a moving target. If a competitor receives approval during your development cycle, the baseline for “substantial improvement” shifts immediately, creating a rescission risk. A strong BTD strategy requires auditing the current standard of care and defining your specific delta: efficacy magnitude, durability, or a subgroup advantage.
Operationalizing BTD: Translating FDA Benefits into Sponsor Action
What BTD Actually Demands From Your Team
A lean biotech team secures BTD for a rare-disease candidate. Their development schedule immediately shifts to high-stakes FDA touchpoints and urgent CMC data requests. BTD advantages only appear if a sponsor is operationally prepared to match the agency’s accelerated pace.
Intensive FDA guidance starting as early as Phase 1 requires immediate alignment on trial design and endpoints — sponsors must defend protocols in real time. Senior FDA management involvement delivers faster issue escalation but demands that internal leadership be available for cross-disciplinary coordination on short notice.
Rolling review readiness allows the FDA to review completed NDA or BLA modules before full submission. This creates a significant documentation burden: CMC, clinical, and non-clinical sections must be finalized and quality-checked months earlier than standard timelines.
BTD success hinges on internal bandwidth and governance. If resourcing is thin, the designation becomes a bottleneck rather than a catalyst.

Timing the BTD Request: Submission Mechanics
While sponsors may submit with the initial IND, the FDA recommends requests no later than the end-of-Phase 2 meeting to maximize development-phase benefits. The agency generally does not accept requests after an original NDA or BLA submission.
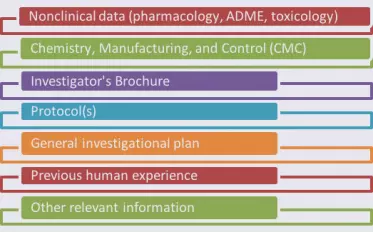
The process is sponsor-initiated. Even if the FDA suggests the pathway during a meeting, the formal submission remains the sponsor’s responsibility. The FDA responds within 60 days of receipt. Place the request in eCTD Module 1, section 1.12.4 (Request for Comments and Advice). Sponsors may also seek preliminary advice before filing to gauge agency sentiment, though this interaction is strictly advisory.
BTD often gets discussed alongside other expedited programs. Choosing the right combination matters.
Mapping FDA Expedited Pathways: When to Stack Them
Most biotech leaders view FDA expedited pathways as a linear progression of prestige, but they are actually modular strategic tools designed to be stacked rather than chosen in isolation. While these programs overlap, they are not interchangeable — success requires understanding the specific evidence thresholds and timing triggers for each.
- Fast Track: Requires preclinical or clinical evidence. Provides frequent FDA access. Available at any stage.
- BTD: Requires preliminary clinical evidence. Provides intensive guidance. Best requested by end-of-Phase 2.
- Accelerated Approval: Uses a surrogate endpoint as the approval mechanism. Typically applies at Phase 2 or 3.
- Priority Review: Requires evidence of significant improvement. Compresses the FDA review clock from ten months to six. Decided at NDA submission.
Strategic stacking is where the real advantage emerges. BTD provides intensive guidance during development, while Priority Review cuts the review timeline. BTD pairs especially well with Accelerated Approval when working with surrogate markers to compress the path to initial commercial launch.
Expedited does not mean simplified. The risks teams underestimate are real.

Managing the Strategic Risks of Breakthrough Programs
The Rescission Risk
Consider a sponsor whose BTD was rescinded because a competitor’s approval redefined the “available therapy” standard. The FDA can withdraw designation if confirmatory data lags or the standard of care shifts during your development cycle.
The CMC Mismatch
Expedited clinical timelines frequently outpace CMC scale-up, stability data, and comparability studies. Parallel CMC planning is critical so that manufacturing hurdles do not become the primary bottleneck for your BLA or NDA submission.
Commercial and Global Considerations
Sponsors must also manage post-approval expectations, including rigorous Postmarketing Requirements (PMRs). A breakthrough label is not an automatic payer win — market access remains driven by clinical evidence quality and label scope. Global programs like EMA PRIME often misalign with FDA timelines, making early international planning critical. While BTD does not grant automatic expanded access, those programs should be evaluated in parallel based on patient need and sponsor capability.
Discuss your cross-functional risk strategy with our team.
How to Assess Your Readiness for Breakthrough Therapy Designation
Securing BTD transforms your development program into an intensive, high-touch partnership with the FDA. This shift requires more than compelling data — it demands a proactive assessment of whether your clinical signals are durable and whether your operations can sustain an accelerated CMC timeline.
Use this decision framework at your next governance meeting to evaluate BTD readiness:
- Indication Depth: Define the condition as life-threatening with a clear gap in the current standard of care.
- Evidence Magnitude: Confirm that preliminary human data demonstrates significant effect size, durability, and endpoint credibility.
- Competitive Narrative: Validate the “substantial improvement” claim against the current, potentially shifting available therapy landscape.
- Trial Flexibility: Confirm the clinical team is prepared to adapt endpoints or designs based on real-time FDA guidance.
- CMC Readiness: Align manufacturing and scale-up plans with a compressed pivotal-to-filing path.
- Submission Infrastructure: Verify that governance and document control systems can support rolling eCTD placement and rapid responses.
BTD increases the frequency of FDA access and the speed of issue escalation. It does not lower the ultimate evidentiary standards for safety or efficacy. If your team is planning an IND amendment or preparing for an end-of-Phase-2 interaction, the right regulatory partnership helps you handle these demands and maximize your designation.
Connect with our regulatory strategists.
Frequently Asked Questions
Does breakthrough therapy designation guarantee FDA approval?
No. BTD is a development-phase accelerator designed to provide more intensive guidance, but the drug must still meet all standard safety and efficacy requirements for an NDA or BLA. BTD reflects the FDA’s assessment of preliminary clinical evidence at a specific point in time. If later trials fail to confirm the initial signal, the agency can deny the final marketing application.
What is the difference between Breakthrough Therapy and Fast Track?
The primary difference lies in the evidence threshold. Fast Track can be granted based on nonclinical or clinical data showing the potential to address an unmet need, while BTD requires preliminary clinical evidence of a substantial improvement over existing treatments. Both offer increased access to the FDA, but BTD provides more intensive involvement from senior agency management throughout the development lifecycle.
How long does the FDA take to respond to a BTD request?
The FDA targets a 60-day response window from receipt of the request. This timeline lets sponsors fold the agency’s decision into immediate clinical and regulatory planning cycles. Plan internal review cycles well before submission to avoid missing the optimal collaboration window.
When is the best time to submit a BTD request?
The FDA recommends submitting no later than the end-of-Phase 2 meeting. This window gives sponsors intensive guidance on pivotal trial design — the program’s most valuable benefit. Requests can be submitted as early as the initial IND, but they are generally not accepted after an original NDA or BLA filing.
Can breakthrough therapy designation be rescinded?
Yes. The FDA can rescind a designation if subsequent data no longer supports the substantial improvement claim. This often happens when larger trials fail to replicate early success or when a newly approved drug changes the standard of care. Keeping evidence generation aligned with the original breakthrough claims throughout development helps mitigate this risk.
Where should the BTD request be placed within the eCTD?
Place the formal request in Module 1 of the eCTD under section 1.12.4 (Request for Comments and Advice). Label the submission clearly so the review team can route it immediately. Include detailed cross-references to clinical evidence in Modules 4 and 5 to help reviewers validate the substantial improvement argument.


{kind=link}