
Throughout the landscape of cancer treatment, timely access to effective therapies is crucial. The FDA’s Real-Time Oncology Review (RTOR) program is a groundbreaking initiative designed to expedite the review process for oncology drugs and biologics, ensuring that patients receive life-saving treatments as quickly as possible.
Why the RTOR Program Was Created
A major challenge with the FDA’s traditional review process is the lengthy review times. Factors that contribute to this challenge include the discovery of unexpected safety and efficacy concerns late in the review cycle and issues in manufacturing, such as quality control lapses or variability in drug production.
In cancer treatment, every day counts. Delays in approval can mean delays in saving lives. Recognizing this urgency, the FDA launched the Oncology Center of Excellence Real-Time Oncology Review (OCE-RTOR) program in 2018. RTOR provides a path to start reviewing critical parts of an application before the full submission is made, reducing overall review time and allowing patients faster access to therapies.
Maintaining and improving review quality while decreasing time-to-approval isn’t an easy task. RTOR addresses this tension by promoting early and standardized data submission, iterative engagement between the FDA and the sponsor, and a more predictable review pathway—without compromising scientific rigor.
RTOR facilitates the early submission of topline efficacy and safety results, prior to the submission of a complete New Drug Application (NDA) or Biologics License Application (BLA), to support an earlier start to the FDA’s evaluation of the application.
Who Is It For and What Does It Cover?
The RTOR program primarily targets therapies intended to treat serious conditions with unmet medical needs—often life-threatening cancers where few or no treatment options exist.
Originally limited to supplemental applications, RTOR has since evolved. It now accepts original NDAs and BLAs for new molecular entities that present a meaningful advantage over existing standards of care. Among the key categories benefiting most are hematologic malignancies (like leukemia) and select solid tumors, especially those with biomarker-driven indications.
Eligibility is generally based on three factors:
- The therapy must demonstrate substantial improvement over existing alternatives, such as increased survival, improved quality of life, or fewer side effects.
- The study designs must be straightforward, with limited complexity to reduce ambiguity during review.
- Endpoints—whether clinical or surrogate—must be clearly interpretable and clinically meaningful.
Importantly, while RTOR accelerates the review timeline, it does not alter the evidence requirements or guarantee final approval. The Prescription Drug User Fee Act (PDUFA) review clock begins only when the full application is received and considered complete by the FDA.
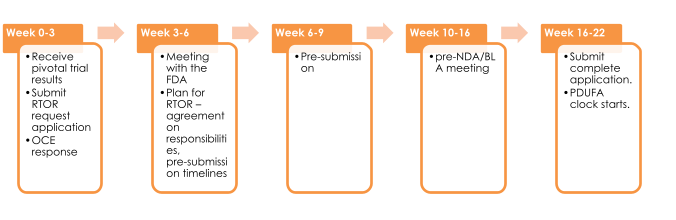
Submissions and Timelines: How It Works
The FDA outlines a flexible roadmap for RTOR participants. Instead of waiting to submit a full application at once—which can easily exceed thousands of pages—sponsors may submit data in staged pre-submissions.
These may include topline safety and efficacy data, proposed labeling, and critical summaries. Early submission helps reviewers begin assessing the benefit-risk profile without delay.
This iterative process allows sponsors and regulators to flag missing data, identify potential issues, and foster communication early—meaning fewer surprises during the full review cycle. The process is especially effective when coupled with other FDA programs like Breakthrough Therapy Designation or Priority Review.
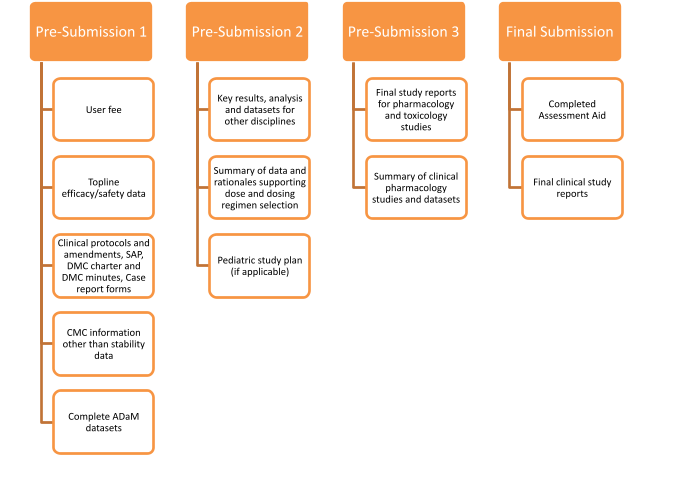
What to Include in Early Submissions
Sponsors can split marketing application content into up to three pre-submissions, followed by a final comprehensive submission. A typical RTOR sequence might include:
- Initial topline clinical results with summary tables or trial datasets
- Draft product labeling and prescribing information
- Integrated summaries of efficacy and safety
- Proposals for Risk Evaluation and Mitigation Strategies (if needed)
This modular approach improves efficiency and helps both FDA and industry allocate resources strategically throughout the review.
CMC Strategy: Keeping Quality Aligned With Speed
Chemistry, manufacturing, and controls (CMC) data are often a bottleneck in regulatory submissions. RTOR’s clinical-aligned timeline requires sponsors to think ahead and tightly align product quality development with clinical milestones.
To fully capitalize on RTOR participation, applicants should begin harmonizing clinical and manufacturing timelines early. Strong CMC planning ensures that when topline clinical data is available, the corresponding quality data—on formulation, stability, manufacturing process, and controls—are equally ready.
Key planning strategies include:
- Proactively communicating with FDA about expected data gaps and development status
- Submitting draft dissolution method development reports for early feedback
- Providing additional real-time stability data or validation batches, even after initial pre-submission
- Arranging early facility readiness for streamlined pre-approval inspections
These steps help reduce risk of post-submission delays often caused by incomplete or unclear CMC packages.
Why RTOR Matters
In fast-moving therapeutic areas like oncology, regulatory innovation is critical. RTOR reflects the FDA’s commitment to modernizing approval pathways, reducing unnecessary delays while upholding scientific standards. By working collaboratively with sponsors via phased submissions, the agency demonstrates a flexible, trust-based model that centers on patient needs.
RTOR’s biggest advantage is information transparency. Reviewing efficacy and safety data early helps the FDA grasp the therapeutic value of a product sooner, surfacing questions about trial design, patient subgroup benefit, statistical methods, or adverse event patterns. This can empower more targeted, data-driven discussions between FDA review teams and sponsors.
Additionally, for sponsors, early feedback can inform commercial planning, real-world evidence strategies, and health technology assessments with payers.
RTOR participation is not without tradeoffs. Sponsors must manage a more complex submission timeline, with frequent document deliveries, iterative agency communication, and the operational burden of having key modules continuously under review.
Smaller biotechs may find resource constraints particularly challenging. Teams must be ready in advance with draft documents, polished analytical reports, and dedicated regulatory personnel to respond to FDA comments in real time.
Despite these considerations, the opportunity to accelerate approval for a life-saving therapy can far outweigh the added effort.

How Syner-G Can Support Your RTOR Journey
Syner-G Biopharma Group can assist with RTOR submissions through our comprehensive regulatory strategy and submission services.
Here are some ways we can help:
- Regulatory Expertise: We have extensive experience in navigating the complexities of regulatory submissions. We provide strategic guidance tailored to the specific requirements of oncology products.
- Document Preparation and Management: We offer meticulous preparation, review, and management of all necessary documents, ensuring they meet the stringent standards of regulatory bodies.
- Global Submission Support: We ensure that submissions are aligned with local and international standards, facilitating a smooth regulatory review process. This includes authoring and reviewing components for various regulatory agencies.
- Project Management: Our project managers coordinate submission timelines and ensure that all interdependent documents and sections are available in a timely manner, preventing delays.
- Regulatory Agency Interaction: We provide support for meetings with regulatory agencies, helping to prepare strategic questions and comprehensive briefing packages, and participating in these crucial interactions.
By leveraging these services, Syner-G Biopharma Group can help streamline the RTOR submission process, potentially accelerating the approval and market readiness of oncology therapies.
Frequently Asked Questions
What does RTOR stand for in oncology?
RTOR stands for Real-Time Oncology Review, an initiative by the FDA’s Oncology Center of Excellence. It allows earlier submission and review of clinical data to speed up cancer treatment approvals.
What is the RTOR oncology review?
RTOR is an FDA review program that lets companies submit cancer drug data early, helping speed up the approval process without compromising safety or quality.
What is the purpose of the RTOR program?
The program aims to shorten review timelines without lowering FDA standards. By reviewing data as it becomes available, the FDA can make faster, informed decisions about oncology therapies.
Who is eligible for RTOR?
RTOR is open to NDAs and BLAs that show clear benefits over existing treatments. Products with straightforward study designs and interpretable endpoints are typically considered.
Does RTOR affect approval timelines?
Yes, RTOR can lead to significantly faster review periods. However, final approval still depends on complete data and regulatory compliance.






{kind=link}